Research
"Charge states of impurity hydrogen in rutile oxide semiconductors"
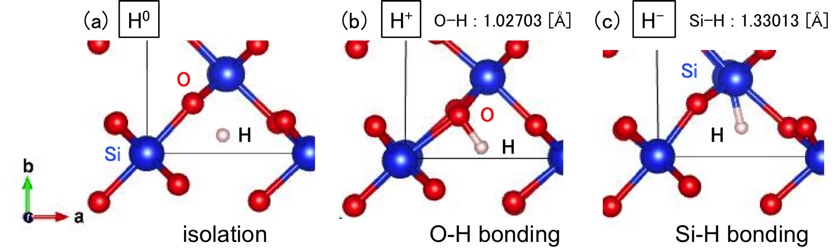
Fig. Charge states of H in SiO2: (a) neutral, (b) positive, and (c) negative.
Impurity hydrogen (H) can take three charge states (H0, H+, H-) and affects the electronic properties of materials. It has been known for passivation of doped semiconductors and semiconductor surfaces, while it is also gathering attention as an electron dopant for oxides where an O2- ion is substituted by a H- ion [1]. Controlling the electrical conductivity is important for the electronic device applications.
We study stability of H in oxide semiconductors with rutile structure MO2 (M=Si, Ge, Sn, Pb, Ti, Zr, Hf) by using first-principles calculation based on density functional theory (DFT). The charge states are determined by Bader analysis [2] after the optimization of SiO2 with a H atom. Figure shows the behavior of H in SiO2. A neutral H atom is localized in interstice as shown in Fig. (a). When H is in H+ state, the H atom is located near the O atom as shown in Fig. (b). The distance between H and O atoms is about 1.03Å, which is almost the same as the O-H bond length in H2O of 0.96Å. Similarly, when H is in H- state in Fig. (c), the H atom forms a Si-H bond of 1.33Å. SiO2 and HfO2 take three charge states (H0, H+, H-), ZrO2 takes H+ and H-, and the others take H+ only. Thus, H exhibits different behavior in these semiconductors.
[1] S. Iimura, S. Matuishi, H. Sato, T. Hanna, Y. Muraba, S.W. Kim, J.E. Kim, M. Takata, and H. Hosono, Nat. Commun. 3, 943 (2012).
[2] R. F. W. Bader, Atoms in Molecules - A Quantum Theory (Oxford University Press, Oxford, 1990).