Research
"Ab initio Prediction of Superconducting Transition Temperature"
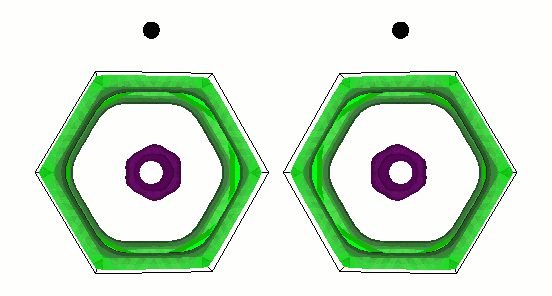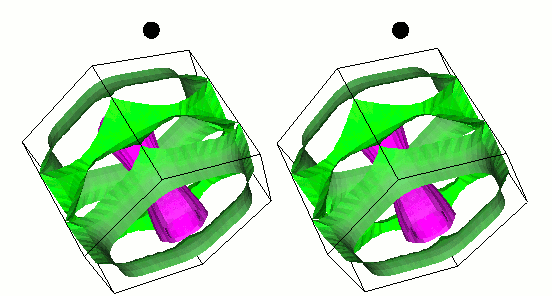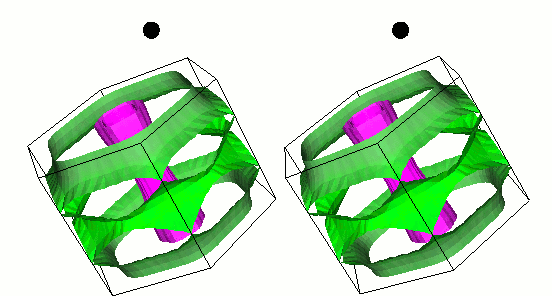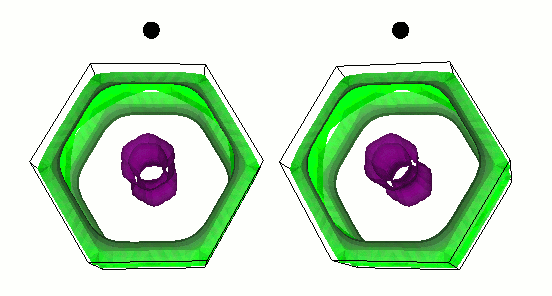
fig. 1: The gap function on the Fermi surface of MgB2. Green (cyan) represents small (large) gap function. It is parallel-eyed stereogram.
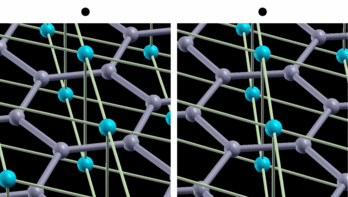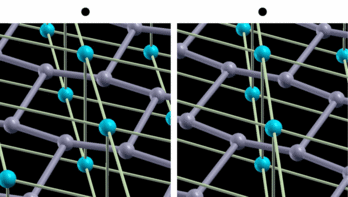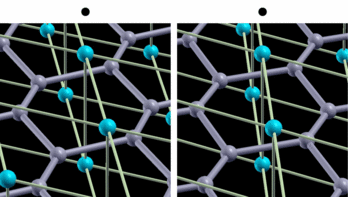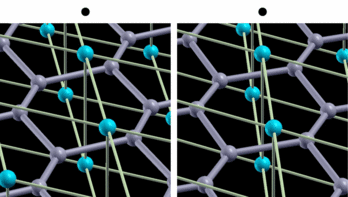
fig. 2: Intra-layer vibrational mode of B atom. Blue (gray) balls represent Mg (B) atoms. It is parallel-eyed stereogram.
Precise prediction of superconducting transition temperature (TC) is one of the grand challenges in first principle calculations. We are developing a numerical code based on density functional theory for superconductors (SCDFT), and applying to a few superconducting materials.
SCDFT is one of extensions of density functional theory (DFT). In the framework of SCDFT, we minimize the free energy as a functional of electron density and a superconducting order parameter. The order parameter obtained by this minimization becomes globally 0 above TC. Moreover, in SCDFT, we can obtain quasi-particle excitation spectra and superconducting gap functions from the non-interacting Kohn-Sham reference system in the self-consistent field.
We show results of an application of our program to magnesium diboride (MgB2), which has the highest TC in phonon-induced type superconductors. Figure 1 shows the magnitude of gap functions on the Fermi surface of MgB2. Cylindrical Fermi surfaces where gap functions are large come from two-dimensional σ-electrons that are localized within the boron layer. And these σ-electrons strongly couple with the intra-layer vibrational mode of the B atom (Fig. 2).