Research
"Pseudopotential Methods"
Pseudopotentials are usually used to reduce computational costs in ab-initio electronic structure calculations, especially in optimizing the structures of solids or calculating the valence band structures of large-scale systems.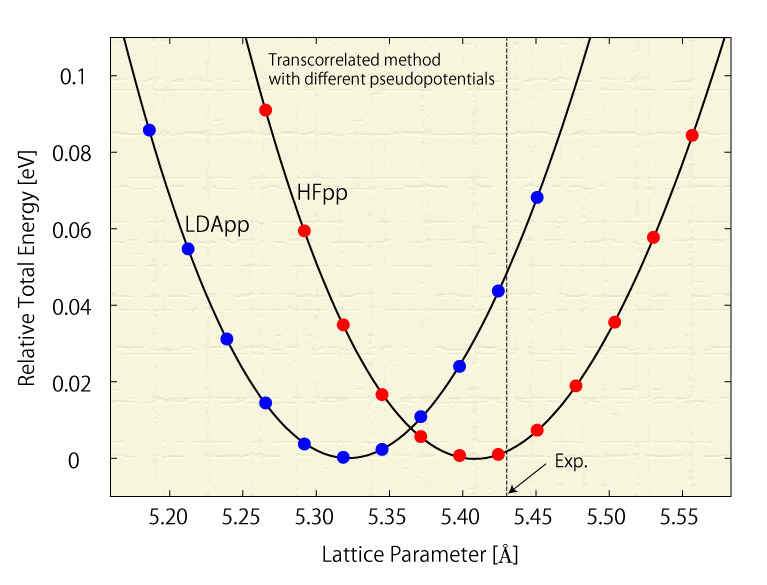 The pseudopotential, consisting of the potential of the nucleus and its core electrons, is dependent on which one-electron approximation is employed, so it is desirable to use pseudopotentials corresponding to each method.
The pseudopotential, consisting of the potential of the nucleus and its core electrons, is dependent on which one-electron approximation is employed, so it is desirable to use pseudopotentials corresponding to each method. We develop a pseudpotential scheme for the transcorrelated method and hybrid-density functional theory methods. The right figure shows the total energy of solid Si calculated by the transcorrelated method with different pseudopotentials, and it illustrates how calculated lattice constants depend on the pseudopotential.
< BACK >