Current Research Areas
In our laboratory, material properties under extreme conditions are studied by utilizing computer simulation methods based on the theory of electronic state, the molecular dynamics, and the first-principle electronic state calculations. The word "first-principle" means solving fundamental equations which rule electrons with basic physical constants such as Planck constants and eliminating empirical parameters. The importance of the first-principle calculation is that it can be applied to the system such as extremely high-pressure condition, complicated bulk surface/interface state, and nanostructures where experimental observations are difficult or sufficient information cannot be obtained by experiments.Here, recent principal research topics are introduced aiming at undergraduate students.
Development of Calculation Methodologies
- Density Functional Theory
- First-Principles Molecular Dynamics
- Path-Integral Molecular Dynamics
- Quantum Monte Carlo
- Transcorrelated Method
- Structural Exploration
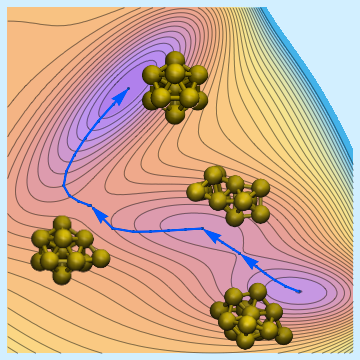
Structural exploration over saddle points of the potential-energy surface
Research topics
"Development of the Transcorrelated
method: wave-function theory for the electronic structure
calculations"
"First-principles calculation of lattice thermal conductivity "
"Pseudopotential Methods"
"Diffusion Monte Carlo method using the transcorrelated trial wave function"
"Development of an efficient Brillouin-zone integration method"
"Structural prediction method by modified potential energy surfaces"
Optical Processes in Solids
- Numerical investigation of the stabilization mechanism of excitonic N-body complexes.
- The amorphization of phase change materials by irradiating ultrashort pulse laser
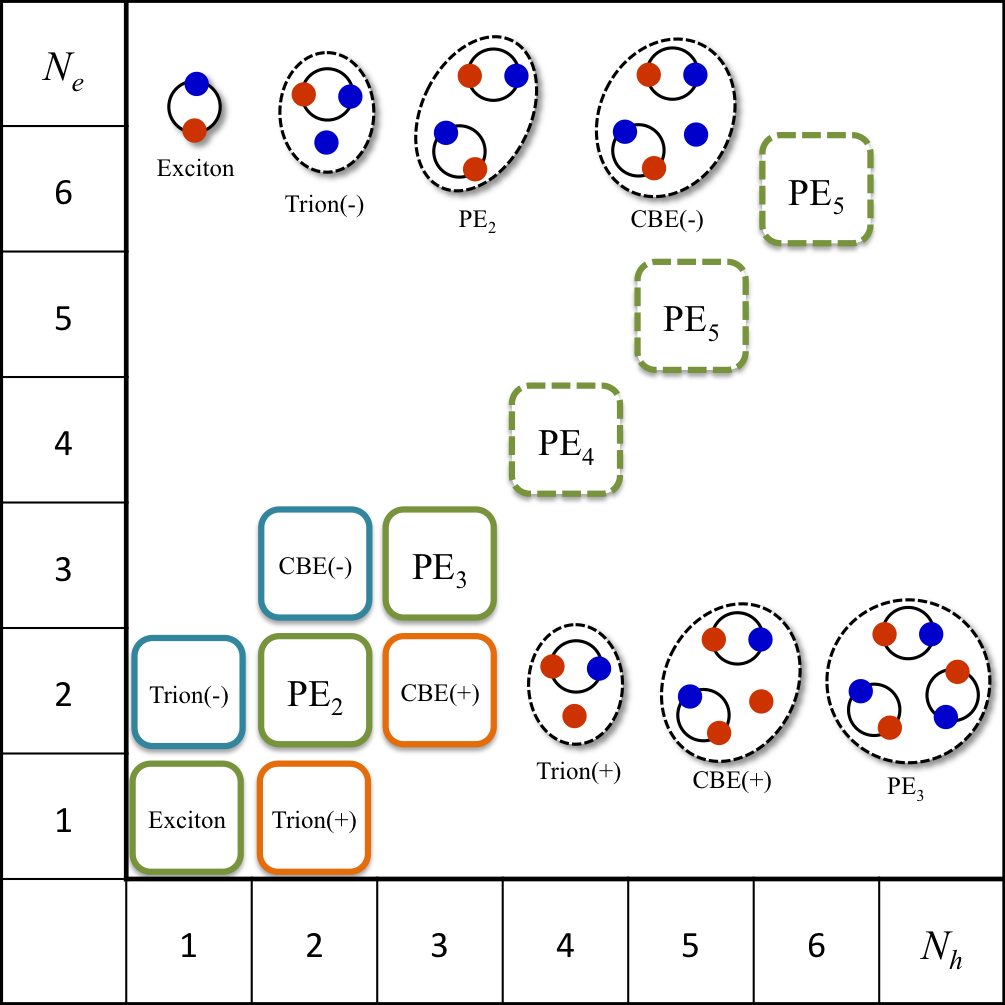
Excitonic complexes in diamond. We showed the stability of complexes circled by solid lines by numerical calculations.
Research topics
"Theoretical investigation of the stability of excitonic N-body complexes in diamond"
"First-principles calculations for amorphization of phase change materials by irradiating ultrashort pulse laser"
Dielectric
- Origin of ferroelectricity
- Properties associated with the vacancy in a dielectric
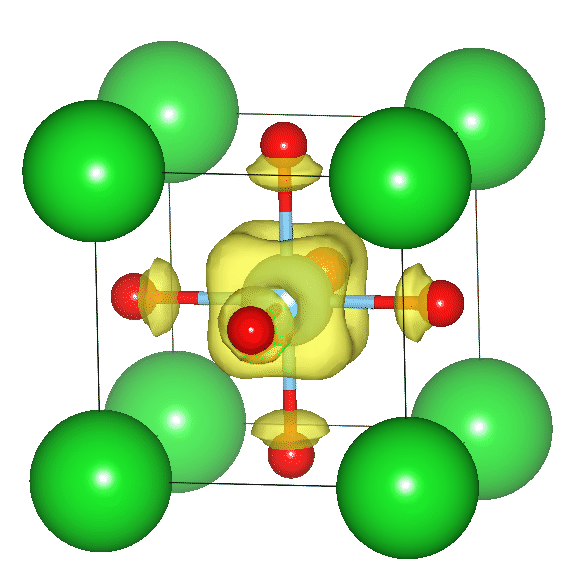
Electron carriers doped in a barium titanate
Research topics
"1H NMR chemical shifts of BaTiO3−xHx"
Physics of Hydrogen in Solids
- Hydrogen in Metals and Semiconductors
- Hydrogen-Bonded Materials
- Solid Hydrogen
- Quantum Effect
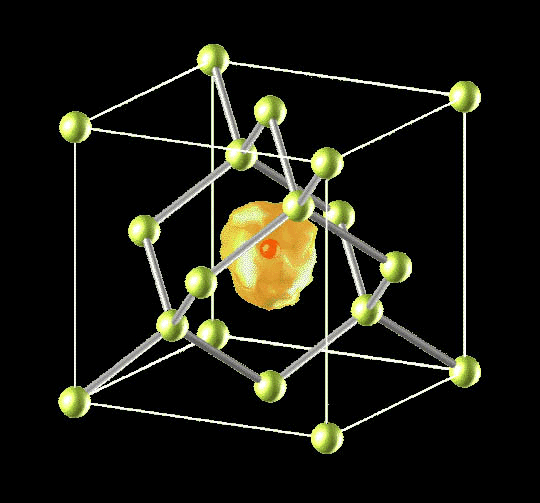
Quantum Distributions of Muon in Muonium State in Crystalline Silicon
Phys. Rev. Lett. 81, 1873-1876 (1998).
Research topics
"Charge states of impurity hydrogen
in rutile oxide semiconductors"
"Prediction of novel perovskite-type oxyhydrides"
Superconductivity
- Ab initio Prediction of Superconducting Transition Temperature
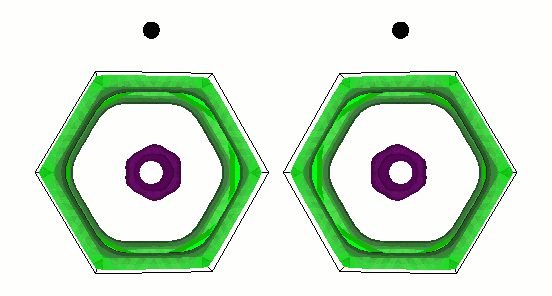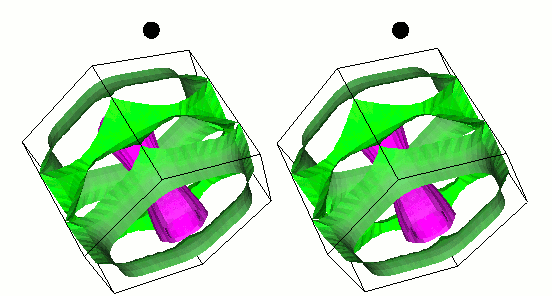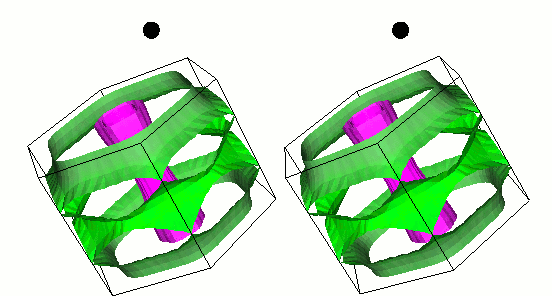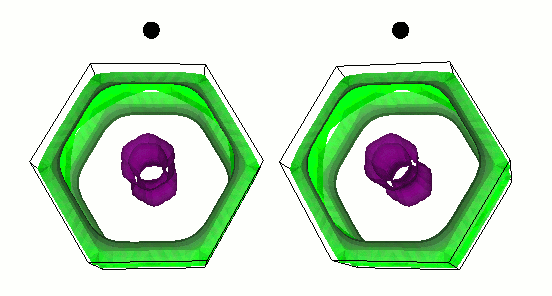
Gap function of MgB2
Research topics
"Ab initio Prediction of
Superconducting Transition Temperature"
"First-principles study on high-temperature superconductivity in hydrides"
Magnetism
- Electronic and spin properties at metal-semiconductor interfaces

Interface structure of Nd magnet
Research topics
"First-principles approach to
grain-boundary structures in Nd magnets"
"Itinerant ferromagnetism at interfaces of nonmagnetic materials"
Thermal Conductivity
- Thermal Conductivity Calculation

Thermal Conductivity Calculation with Non-Equilibrium Molecular Dynamics
Research topics
"Lattice Thermal-Conductivity
Calculated with Anharmonic Lattice Model"
"Thermal conductivity reduction by rattling vibrations"
Surface and Interface Science
- Electronic and spin properties at metal-semiconductor interfaces
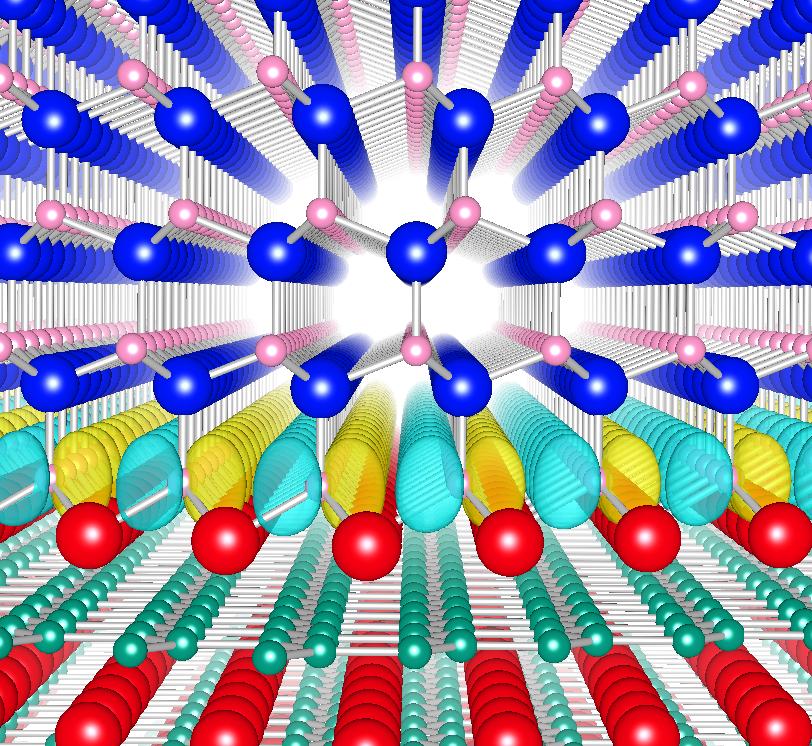
Spin-polarized wave function at the AlN/MgB2 interface
Phys. Rev. Lett. 106, 047201 (2011).
Research topics
"Itinerant ferromagnetism at
interfaces of nonmagnetic materials"
"Electronic properties of SiON/SiC(0001) surface ~ band-gap profile, Schottky contact ~"
"Property of organic molecules well arranged on a semi-conductor surface"
"First-principles study of Cu(100)-c(2x2)N surface"
High-Pressure Physics
- Phase Transition under High Pressure
- Mineral Properties
- High-Pressure Synthesis

Nuclei distribution of solid hydrogen under high pressure
Nature 404, 259-262 (2000).