研究紹介
「第一原理計算によるCu(100)-c(2×2)N表面の研究」
第一原理計算によってCu(100)-c(2×2)N表面を研究した。 この表面では適切なN原子の被覆率で約5nm四方のc(2×2)N 構造の島がCu清浄面で区切られた正方格子を組む。 この自己組織化構造は磁性物質を整列させるナノサイズのテンプレートとして使われている。 この系について最近の実験によって2回対称の局所構造が、 通説であった4回対称の構造の代りに提案されていたが、 最適化されたCu(100)-c(2×2)N表面の構造は4回対称性を持っていた。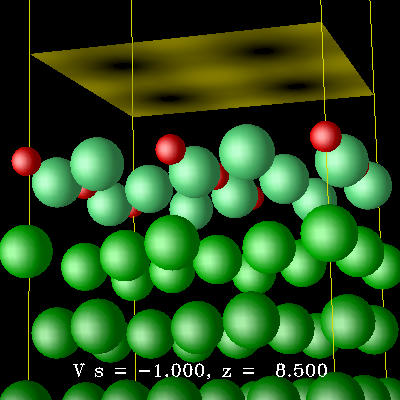 シミュレートされたSTM像は通説での解釈を支持した。
また自己組織化を誘導している力を理解するためにN原子間の有効相互作用エネルギーを求めた。
さらにN原子の拡散バリヤも求めた。
これらの計算のためにSCF iterationの前処理法を一つ開発して、
計算時間を短縮した。
シミュレートされたSTM像は通説での解釈を支持した。
また自己組織化を誘導している力を理解するためにN原子間の有効相互作用エネルギーを求めた。
さらにN原子の拡散バリヤも求めた。
これらの計算のためにSCF iterationの前処理法を一つ開発して、
計算時間を短縮した。 図はCu(100)-c(2x2)N表面のSTM像のシミュレーション。 STM像で明るいのは赤いN原子である。 Cu原子は緑色であり、明るい緑のものほど表面に近い。 (クリックすると拡大します)
[1] Y. Yoshimoto and S. Tsuneyuki, "First principles study of
inter-nitrogen interaction energy of Cu(100)-c(2 x 2)N surface",
Int. J. Quant. Chem. 91, 211-215 (2003).
[2] Y. Yoshimoto and S. Tsuneyuki, "First-principles study of inter nitrogen interaction energy of Cu(100)-c(2 x 2)N surface",
Surf. Sci. 514,
200-205 (2002).
< BACK >